Oral Anticoagulants — NOACs, Antiplatelets & Fibrinolytics
Heparins, Warfarin, DOACs, Antiplatelet Drugs, Fibrinolytics & Reversal Agents
Past RGUHS + DNB + MPMSU + MUHS + VNSGU · 49
MPMSUOct '25
MPMSUJan '25
RGUHSJun '24
RGUHSJun '24
MUHSWinter '24
MPMSUJun '23
DNBOct '23
VNSGUJun '23
RGUHSNov '22
RGUHSMay '22
RGUHSJul '21
MPMSUAug '21
DNBDec '21
MUHSSummer '21
RGUHSNov '20
MPMSUJul '20
DNBJun '20
RGUHSNov '19
RGUHSMay '19
MPMSUMay '19
MPMSU2019
MUHSSummer '19
MUHSWinter '19
RGUHSMay '18
RGUHSMay '18
RGUHSMay '18
MPMSUMay '18
MPMSUMay '18
MPMSU2017
MPMSUJun '17
MUHSSummer '17
MUHSSummer '16
VNSGUApr '16
MPMSU2014
DNBDec '14
DNBDec '14
MUHSWinter '14
DNBDec '13
DNBDec '12
RGUHSMay '11
MPMSU2011
MPMSU2011
DNBDec '11
RGUHSOct '10
RGUHSMay '09
RGUHSOct '08
RGUHSApr '08
RGUHSApr '06
MPMSU2004
Oral Anticoagulants, NOACs, Antiplatelets & Fibrinolytics
1. Haemostasis, the coagulation cascade & natural anticoagulants
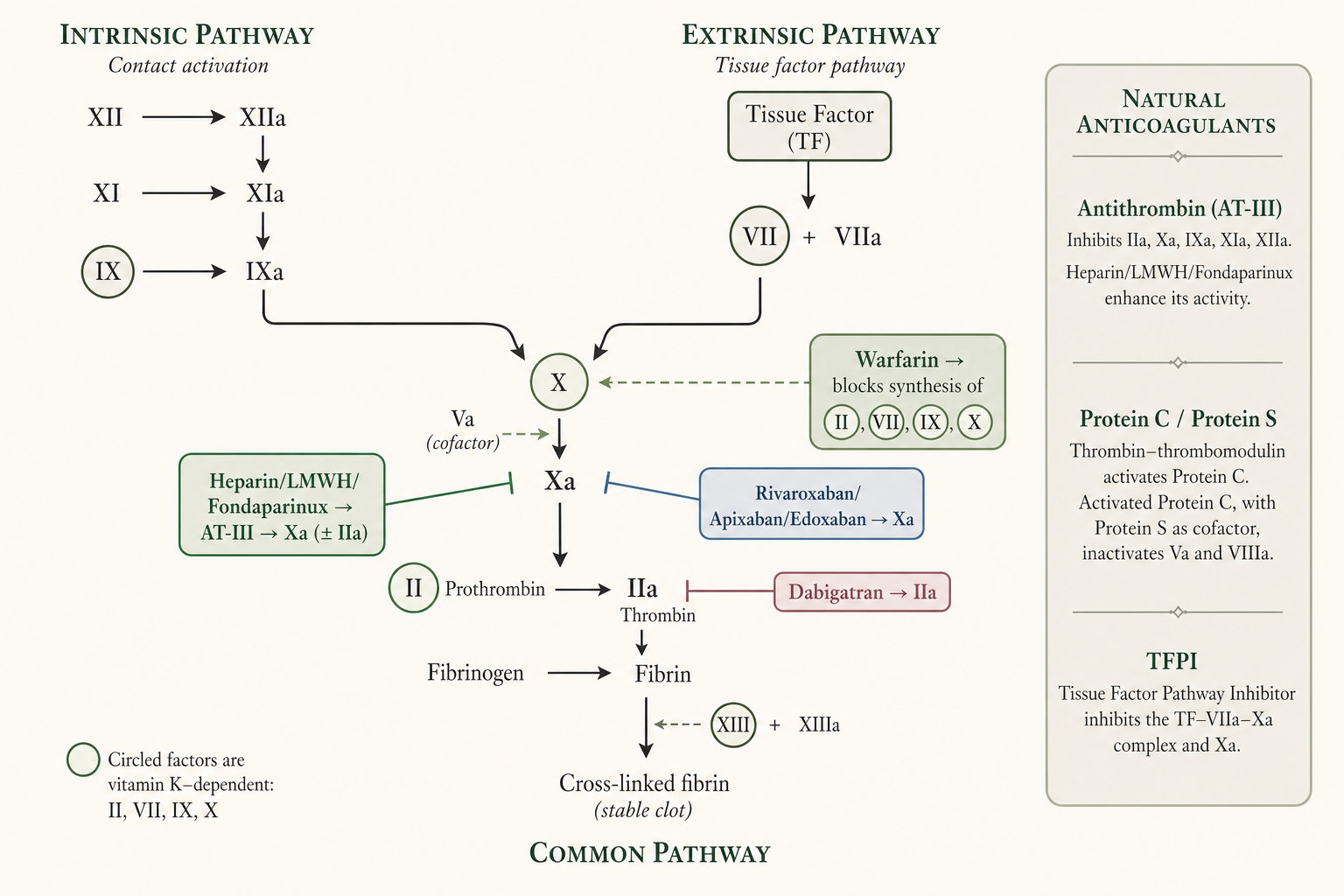
- Haemostasis is the finely regulated arrest of blood loss from a damaged vessel while blood remains fluid within the intact vasculature; a delicate balance between coagulation and fibrinolysis normally prevents both thrombosis and haemorrhage (G&G 14e Ch.36, pp.709–10).
- Three sequential events: (i) platelet adhesion to subendothelial macromolecules → activation; (ii) release of mediators that recruit/activate nearby platelets → aggregation to form the primary haemostatic plug; (iii) vessel-wall injury exposes tissue factor (TF) which initiates coagulation, generating a burst of thrombin (factor IIa) that converts fibrinogen → fibrin, stabilising the plug (G&G 14e Ch.36, p.709; Katzung 16e Ch.34, pp.640–1).
- Conversion of fibrinogen to fibrin: fibrinogen is a 340-kDa dimer of three paired chains (Aα, Bβ, γ). Thrombin releases fibrinopeptide A (16-aa) and fibrinopeptide B (14-aa) from the amino-termini, creating "knobs" that fit "holes" on other monomers → a non-covalent fibrin gel (the end-point of in-vitro clotting tests). Factor XIIIa (a thrombin-activated transglutaminase) then covalently cross-links adjacent monomers to strengthen the clot (G&G 14e Ch.36, p.710).
- Vitamin K–dependent factors = II (prothrombin), VII, IX, X (plus anticoagulant proteins C, S, Z). 9–13 amino-terminal Glu residues are γ-carboxylated to Gla (γ-carboxyglutamic acid) post-translationally; Gla residues bind Ca2+ and anchor the factors to the anionic phospholipid membrane of activated platelets — essential for coagulant activity (G&G 14e Ch.36, p.710; KDT 8e Ch.45, p.661).
- Non-enzymatic cofactors: TF (constitutively on subendothelial smooth muscle/fibroblasts, exposed on injury; binds factor VIIa), factor V and factor VIII (procofactors). Thrombin activates V→Va and VIII→VIIIa; VIIIa is the receptor for IXa, Va is the receptor for Xa on the activated platelet surface (G&G 14e Ch.36, p.710).
- Prothrombinase complex (Xa + Va + anionic phospholipid + Ca2+) activates prothrombin 109-fold more efficiently than factor Xa alone (G&G 14e Ch.36, p.711).
- Initiation — extrinsic (TF/VIIa) pathway: TF–VIIa, in the presence of anionic phospholipid and Ca2+ (extrinsic tenase), shows 30,000-fold increased activity over VIIa alone; activates X and IX. This is the main physiological initiator in vivo (G&G 14e Ch.36, p.711; Katzung 16e Ch.34, p.642).
- Intrinsic (contact) pathway: factor XII + prekallikrein + HMW kininogen on a negatively charged surface (kaolin, glass, also cell-free DNA, NETs, platelet polyphosphates) → XIIa → XIa → IXa. Intrinsic tenase (IXa + VIIIa + phospholipid + Ca2+) activates X more efficiently than TF–VIIa, so optimal thrombin generation depends on it (G&G 14e Ch.36, p.711).
- Clinically silent contact factors: deficiency of XII, prekallikrein or HMW kininogen does NOT cause bleeding; factor XI deficiency = mild/variable bleeding; factor VIII or IX deficiency = haemophilia A or B (spontaneous, potentially fatal bleeding) (G&G 14e Ch.36, pp.711–12; Katzung 16e Ch.34, p.642).
- In-vitro tests (KDT framing): aPTT tests intrinsic + common pathway (normal 26–32 s; intrinsic-pathway defect → prolonged aPTT, normal PT); PT tests extrinsic + common pathway (normal 12–14 s; extrinsic defect → prolonged PT, normal aPTT); common-pathway defect → both prolonged (KDT 8e Ch.45, p.659).
- Natural anticoagulant mechanisms (4): (i) endothelial NO + prostacyclin (PGI2) + CD39 inhibit platelet activation; (ii) antithrombin (AT/AT-III) — a serpin that "suicide-substrate" inhibits thrombin, Xa (and IXa, XIa, XIIa); heparan sulfate enhances it ~1000-fold; (iii) protein C — bound to endothelial protein C receptor (EPCR), activated by thrombin–thrombomodulin, then with cofactor protein S degrades Va and VIIIa; (iv) tissue factor pathway inhibitor (TFPI) — binds Xa, then the binary complex inhibits TF-bound VIIa (G&G 14e Ch.36, p.712; Katzung 16e Ch.34, p.643).
- Arterial vs venous thrombi (therapeutic corollary): arterial = platelet-rich "white" thrombi (high shear) → antiplatelet drugs more useful; venous = fibrin-rich "red" thrombi with trapped RBCs (sluggish flow, "red tail") → anticoagulants more useful (KDT 8e Ch.45, p.676; Katzung 16e Ch.34, pp.641–2).
- Factor V Leiden (resistance to inactivation by protein C/S) is the commonest inherited thrombophilia; deficiencies of AT, protein C or protein S also raise venous-thrombosis risk; antiphospholipid antibody syndrome is a key acquired risk factor (Katzung 16e Ch.34, pp.643, 653).
Continue reading
Oral Anticoagulants Noacs
PharmaNotes Pro · Comprehensive
Sign in with your Google account. If you're already subscribed, the chapter unlocks immediately — otherwise, pick Monthly or Annual on the next step.